Newsroom
Acceleration of fragment-based drug discovery through AI and language models
EU-OPENSCREEN ERIC leads Fragment-Screen work package 6 (WP6) on AI-supported fragment-to-lead optimisation, aiming to bridge the gap between experimental and computational approaches in de novo drug discovery. Under the co-lead of Prof Tiago Rodrigues of EU-OPENSCREEN partner site Faculty of Pharmacy, University of Lisbon (FFUL) and research scientist Jannis Born of Fragment-Screen partner IBM Research, the four WP6 partners [6] are devising a design–make–test–analyse (DMTA) cycle (see Figure 1) that utilises generative AI developed by researchers at IBM Research Europe [7].
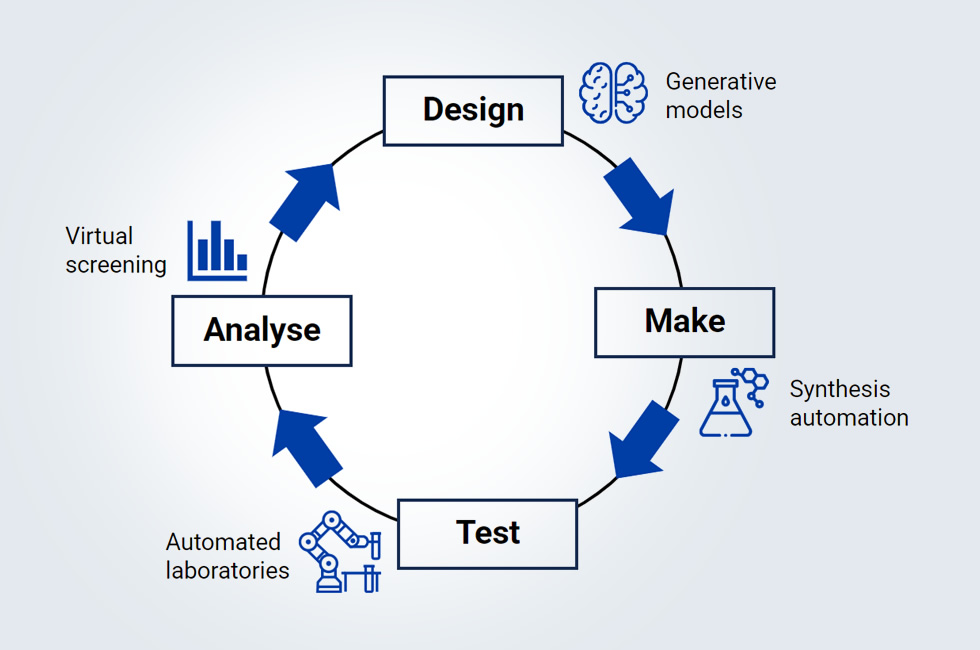
This generative AI leverages chemical language models that are powered by the same principles underlying today's large language models (LLMs). The AI suggests fragments for experimental synthesis and wet-lab validation to assess potential binding to a given protein target. The drug candidates are designed in a two-step process. Initially, promising or user-defined chemical motifs are combined to form small novel scaffolds. A second AI, called Regression Transformer [8], then decorates these scaffolds and identifies the most potent fragments from a vast array of structural analogues of each initial scaffold. For an informed design, the AI uses not only information from existing ligands (if available) but can also be conditioned on different parameters of interest, such as predicted affinity against the target, ligand efficiency, or optimal physicochemical properties.
The medicinal chemistry researchers working on different protein targets have significant flexibility in how much control they retain over the design process. For instance, if chemists have specific ideas about the presence or absence of certain motifs in the final molecule, the generative AI can constrain its design space to molecules that include or exclude such motifs. Conversely, chemists can use the same pipeline in an unconstrained manner, allowing the AI to initiate the design from a ‘blank canvas’.
Currently, WP6 partners are exploring this concept for the design of ligands against four targets of SARS-CoV-2: (1) the main protease (MPro/nsp5), (2) the macrodomain (nsp3b), (3) the nucleocapsid protein, and (4) the nsp10–nsp16 complex. Starting with the structural data collected in the COVID Moonshot program [9] – accessible through the Fragalysis webserver [10] – the projects began by prioritising fragments for medicinal chemistry elaboration. Since different protein targets have been studied to varying extents, collecting sufficient data to train the AI models was a significant challenge for some projects. This hurdle was addressed by supplementing the Fragalysis data with existing public datasets, generating synthetic data, or acquiring new data experimentally.
Within the first six months of research, IBM Research successfully generated novel potentially active molecules for each of the four targets. Currently, medicinal chemistry sites are synthesising these molecules, through the support of IBM’s AI chemical synthesis platform RXN for Chemistry [11], for automated synthesis planning. Experimental validation will follow and may include profiling of the molecules via surface plasmon resonance (SPR), nuclear magnetic resonance (NMR), or X-ray crystallography. The DMTA cycle will then be completed by incorporating the freshly generated data points into the deep learning models, thereby refining the generative AI and facilitating the design of another round of increasingly tailored chemical entities.
This update was written by Tiago Rodrigues of the University of Lisbon and Jannis Born of IBM Research Europe and reviewed by Tanja Miletic and Alexandra Ertman of EU-OPENSCREEN.


[1] https://fragmentscreen.org/
[2] https://www.eu-openscreen.eu/
[4] https://elixir-europe.org/
[5] https://instruct-eric.org/
[6] Fragment-Screen WP6 partners: i) Medicinal chemistry groups: Center for Biological Research Margarita Salas, CSIC, Spain (EU-OPENSCREEN), University of Lisbon, Research Institute for Medicine, Portugal (EU-OPENSCREEN), Latvian Institute of Organic Synthesis, Latvia (EU-OPENSCREEN), Technical University of Denmark, Denmark; ii) structural biology groups: Goethe University Frankfurt, Germany; XChem group, Diamond Light Source, UK; Netherlands Cancer Institute, the Netherlands; ii) AI experts: IBM Research Europe.
[7] https://www.zurich.ibm.com/
[8] https://www.nature.com/articles/s42256-023-00639-z
[9] https://www.nature.com/articles/s41557-020-0496-2#citeas
[10] https://fragalysis.diamond.ac.uk/